autor: mgr Klaudia Sałamunia
Zespół Wiskotta-Aldricha (WAS, Wiskott-Aldrich syndrome) jest rzadkim, pierwotnym niedoborem odporności sprzężonym z chromosomem X. Choroba została po raz pierwszy opisana przez Alfreda Wiskotta w 1937 roku, natomiast rodzinne występowanie schorzenia i sposób dziedziczenia wykazał Robert Aldrich w 1954 roku. WAS charakteryzuje się klasyczną triadą objawów: małopłytkowością z mikrotrombocytopenią, egzemą oraz nawracającymi zakażeniami. Współcześnie wiadomo jednak, że obraz kliniczny choroby jest znacznie bardziej złożony i obejmuje również autoimmunizację, przewlekły stan zapalny oraz zwiększoną predyspozycję do nowotworów układu chłonnego i krwiotwórczego.
Częstość występowania szacuje się na około 1 : 100 000 żywych urodzeń (głównie męskich) bez wyraźnych predyspozycji etnicznych lub geograficznych.
Patogeneza zespołu związana jest z mutacjami genu WAS zlokalizowanym na ramieniu krótkim chromosomu X (Xp11.23), który koduje białko WASp. Opisano dotychczas ponad 300 mutacji (m.in. delecje, insercje oraz mutacje punktowe) prowadzące do zaburzenia lub braku ekspresji białka (ang. LOF - loss of function). Białko WASp obecne jest w komórkach hematopoetycznych, limfocytach T i B, makrofagach oraz płytkach krwi. Jego główną funkcją jest aktywacja polimeryzacji rozgałęzionych włókien aktyny, co umożliwia komórkom zmianę kształtu, migrację i tworzenie synaps immunologicznych.
Upośledzenie funkcji białka wiąże się z zaburzeniem reorganizacji cytoszkieletu aktynowego i upośledzeniem komunikacji międzykomórkowej układu odpornościowego. Przekłada się to na: dysfunkcję limfocytów T, zaburzenia odpowiedzi humoralnej, defekty komórek NK, nieprawidłową fagocytozę, zwiększoną apoptozę komórek hematopoetycznych i nieprawidłową produkcję i przeżycia płytek krwi. U około 25-70% pacjentów dochodzi do autoimmunizacji najczęściej w postaci niedokrwistości autoimmunohemolitycznej, zapalenia naczyń, neutropenii autoimmunologicznej, pierwotnej immunologicznej małopłytkowości, zapalenia jelit.
Przez wiele lat rokowanie było niekorzystne — większość pacjentów umierała przed 10. rokiem życia z powodu zakażeń lub krwawień. Współczesne leczenie znacząco poprawiło przeżycie chorych. Istotnym problemem klinicznym jest zwiększona predyspozycja do nowotworów układu chłonnego i krwiotwórczego, głównie chłoniaków i białaczek. U części pacjentów rozwijają się także choroby autoimmunologiczne oraz nefropatie związane z odkładaniem kompleksów immunologicznych. Badania wykazały dużą zmienność fenotypową choroby — tylko część pacjentów prezentuje pełną klasyczną triadę objawów. U niektórych dominującym objawem pozostaje małopłytkowość przy łagodniejszym przebiegu niedoboru odporności. Rozpoznanie opiera się na obrazie klinicznym, badaniach immunologicznych oraz diagnostyce genetycznej. Ważną metodą leczenia jest przeszczepienie szpiku kostnego, a współcześnie rozwijana jest również terapia genowa.
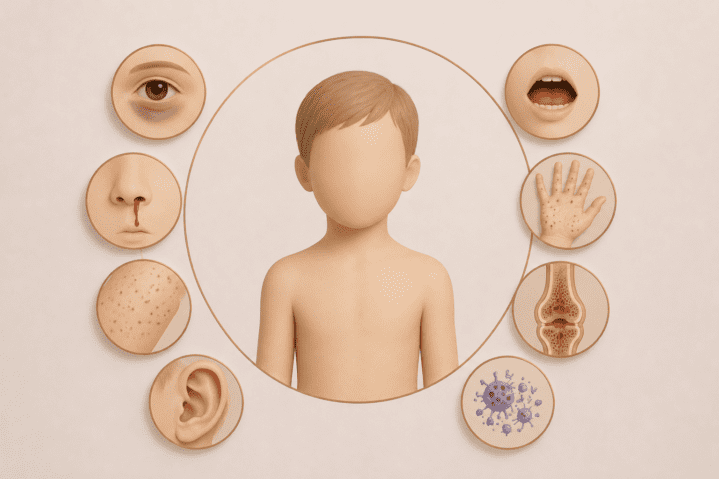 Obraz fenotypowy zespołu można rozdzielić na 3 rodzaje objawów: hematologiczne, immunologiczne oraz dermatologiczne. Pierwsze z nich to przede wszystkim małopłytkowość skutkująca wybroczynami, krwawieniem z nosa, krwawymi biegunkami, krwawieniem śródczaszkowym oraz łatwym siniaczeniem. Objawy immunologiczne związane są z nawracającymi infekcjami i zakażeniami (typowe są tu zapalenia ucha, zapalenia płuc, infekcje skóry, zakażenia HSV i EBV). Ostatni rodzaj wiąże się z ciężkimi wypryskami atopowymi i przewlekłymi zmianami skórnymi. Charakterystyczną cechą hematologiczną WAS jest obecność małych płytek krwi (mikrotrombocytopenia). U części kobiet będących nosicielkami mutacji obserwowano również obniżoną liczbę płytek krwi.
Obraz fenotypowy zespołu można rozdzielić na 3 rodzaje objawów: hematologiczne, immunologiczne oraz dermatologiczne. Pierwsze z nich to przede wszystkim małopłytkowość skutkująca wybroczynami, krwawieniem z nosa, krwawymi biegunkami, krwawieniem śródczaszkowym oraz łatwym siniaczeniem. Objawy immunologiczne związane są z nawracającymi infekcjami i zakażeniami (typowe są tu zapalenia ucha, zapalenia płuc, infekcje skóry, zakażenia HSV i EBV). Ostatni rodzaj wiąże się z ciężkimi wypryskami atopowymi i przewlekłymi zmianami skórnymi. Charakterystyczną cechą hematologiczną WAS jest obecność małych płytek krwi (mikrotrombocytopenia). U części kobiet będących nosicielkami mutacji obserwowano również obniżoną liczbę płytek krwi.
Leczenie zespołu Wiskotta–Aldricha (WAS) obejmuje zarówno terapię wspomagającą, jak i leczenie przyczynowe. Postępowanie konwencjonalne koncentruje się na profilaktyce i leczeniu zakażeń przy użyciu antybiotyków o szerokim spektrum działania, leków przeciwwirusowych oraz przeciwgrzybiczych, a także na zapobieganiu powikłaniom krwotocznym poprzez transfuzje koncentratu płytek krwi. W terapii zmian skórnych o charakterze egzemy stosuje się miejscowe glikokortykosteroidy. U pacjentów z istotnym niedoborem przeciwciał zalecana jest dożylna lub podskórna terapia immunoglobulinami (IVIG/SCIG), przy czym dawki immunoglobulin są zwykle wyższe niż w innych pierwotnych niedoborach odporności ze względu na zwiększony katabolizm immunoglobulin obserwowany w WAS. W wybranych przypadkach stosuje się eltrombopag – agonistę receptora trombopoetyny – w celu ograniczenia ryzyka krwawień, zwłaszcza u pacjentów oczekujących na przeszczepienie krwiotwórczych komórek macierzystych. Leczenie immunosupresyjne, w tym rytuksymab, znajduje zastosowanie u chorych z objawami autoimmunologicznymi, szczególnie w przebiegu cytopenii autoimmunologicznych. U części pacjentów rozważa się splenektomię, która może prowadzić do zwiększenia liczby płytek krwi i zmniejszenia skłonności do krwawień, jednak wiąże się ze zwiększonym ryzykiem ciężkich zakażeń i koniecznością długotrwałej profilaktyki antybiotykowej. Obecnie jedyną uznaną metodą leczenia o charakterze potencjalnie curatywnym pozostaje przeszczepienie krwiotwórczych komórek macierzystych (HSCT/HCT), szczególnie skuteczne w przypadku zgodności HLA dawcy i biorcy. Alternatywną, intensywnie badaną strategią terapeutyczną jest terapia genowa, stanowiąca obiecującą opcję leczenia przyczynowego u pacjentów z WAS.
Podsumowując, Zespół Wiskotta-Aldricha jest złożoną chorobą genetyczną obejmującą zarówno układ odpornościowy, jak i krwiotwórczy. Mimo że klasycznie kojarzony jest z triadą objawów obejmującą małopłytkowość, egzemę i nawracające zakażenia, współczesne badania pokazują, że przebieg choroby może być bardzo różnorodny i obejmować także autoimmunizację, przewlekły stan zapalny oraz zwiększoną predyspozycję do nowotworów hematologicznych. Postęp diagnostyki molekularnej pozwolił lepiej zrozumieć rolę mutacji genu WAS oraz mechanizmy odpowiedzialne za zmienność fenotypową choroby. Jednocześnie rozwój nowoczesnych metod leczenia, szczególnie przeszczepiania krwiotwórczych komórek macierzystych i terapii genowej, znacząco poprawił rokowanie pacjentów oraz otworzył nowe możliwości terapii chorób genetycznych układu immunologicznego.
Piśmiennictwo:
Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kohn DB. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15(1 Suppl):84–90.
Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125(6 Pt 1):876–885.
Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. Curr Opin Hematol. 2008;15(1):30–36.
Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat Rev Immunol. 2010;10(3):182–192.
Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78(4):635–644.
Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. 2013;1285:26–43.
Ferrua F, Cicalese MP, Galimberti S, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6(5)–e253.
Albert MH, Bittner TC, Nonoyama S, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. 2010;115(16):3231–3238.
Worth AJJ, Thrasher AJ. Current and emerging treatment options for Wiskott-Aldrich syndrome. Expert Rev Clin Immunol. 2015;11(9):1015–1032.