autor: mgr Klaudia Sałamunia
Zespół Waardenburga jest rzadkim schorzeniem genetycznym, najczęściej dziedziczonym autosomalnie dominująco, wynikającym z zaburzeń migracji i proliferacji komórek grzebienia nerwowego podczas embriogenezy. Choroba została po raz pierwszy opisana w 1951 roku przez Petrus Johannes Waardenburg. Nieprawidłowy rozwój melanocytów prowadzi do zaburzeń pigmentacji skóry, włosów i tęczówek oraz do wrodzonego niedosłuchu odbiorczego.
Częstość występowania na świecie szacuje się na 1 przypadek na 42 000–50 000 osób. Stanowi najczęstszą przyczynę autosomalnie dominującego zespołowego niedosłuchu, odpowiadając za około 2–5% przypadków wrodzonej głuchoty oraz za 0,9–2,8% przypadków w populacji osób niesłyszących. Występuje we wszystkich grupach etnicznych i rasowych, bez wyraźnych różnic w zależności od płci. Obserwuje się regionalne różnice w częstości występowania, m.in. wyższą zapadalność w populacji kenijskiej oraz niższe wartości odnotowane historycznie w Holandii, co wiązano z niską penetracją fenotypową choroby. Jako wrodzone zaburzenie dziedziczne, zespół Waardenburga jest zwykle rozpoznawany już w okresie noworodkowym lub we wczesnym dzieciństwie, jednak objawy dermatologiczne mogą wykazywać zmienną ekspresję w ciągu życia pacjenta.
Zespół Waardenburga jest chorobą uwarunkowaną genetycznie, najczęściej dziedziczoną autosomalnie dominująco, której patogeneza związana jest z zaburzeniami rozwoju komórek grzebienia nerwowego podczas embriogenezy. Mutacje obejmujące insercje, delecje, zmiany sensu, nonsensu oraz przesunięcia ramki odczytu prowadzą do nieprawidłowej migracji, proliferacji i różnicowania melanoblastów oraz innych komórek pochodzących z grzebienia nerwowego. Skutkiem tych zaburzeń są charakterystyczne nieprawidłowości pigmentacyjne skóry, włosów i tęczówek, a także wrodzony niedosłuch odbiorczy wynikający z nieprawidłowego rozwoju struktur ucha wewnętrznego.
Kluczową rolę w patogenezie zespołu odgrywają geny regulujące rozwój melanocytów i komórek układu nerwowego, przede wszystkim PAX3, MITF, SOX10, EDNRB oraz EDN3. Gen PAX3 koduje czynnik transkrypcyjny niezbędny dla prawidłowego rozwoju struktur wywodzących się z grzebienia nerwowego i odpowiada za większość przypadków typu 1 i 3 zespołu. Z kolei mutacje genu MITF, regulującego dojrzewanie i funkcję melanocytów, są najczęściej związane z typem 2 choroby. Mutacje genów EDNRB, EDN3 i SOX10 zaburzają rozwój zarówno melanocytów, jak i zwojów jelitowych, co tłumaczy współwystępowanie choroby Hirschsprunga w typie 4 zespołu Waardenburga.
Wyróżnia się cztery główne typy kliniczne zespołu Waardenburga. Typ 1, związany z mutacjami genu PAX3, charakteryzuje się obecnością dystopii canthorum, szeroką nasadą nosa, zaburzeniami pigmentacji oraz wrodzonym niedosłuchem odbiorczym. Typ 2 wykazuje podobny obraz kliniczny, jednak bez dystopii canthorum; najczęściej związany jest z mutacjami genu MITF. Typ 3, określany również jako zespół Kleina-Waardenburga, stanowi cięższą odmianę typu 1 i obejmuje dodatkowo wady układu mięśniowo-szkieletowego, zwłaszcza kończyn górnych. Typ 4, zwany zespołem Waardenburga-Shaha, współistnieje z chorobą Hirschsprunga i najczęściej dziedziczy się autosomalnie recesywnie.
| Typ | Uszkodzony gen | Sposób dziedziczenia | Charakterystyczne cechy kliniczne | Cechy różnicujące |
| Typ 1 (WS1) | PAX3 | Autosomalny dominujący | Niedosłuch odbiorczy, heterochromia tęczówek, biała grzywka, plamista depigmentacja skóry, szeroka nasada nosa | Obecność dystopii canthorum |
| Typ 2 (WS2) | MITF, SOX10, rzadziej inne | Autosomalny dominujący | Niedosłuch odbiorczy, zaburzenia pigmentacji włosów, skóry i tęczówek | Brak dystopii canthorum |
| Typ 3 (WS3, zespół Kleina-Waardenburga) | PAX3 | Autosomalny dominujący | Objawy typu 1 oraz wady układu mięśniowo-szkieletowego | Nieprawidłowości kończyn górnych (przykurcze, hipoplazja mięśni, zrosty) |
| Typ 4 (WS4, zespół Waardenburga-Shaha) | EDNRB, EDN3, SOX10 | Najczęściej autosomalny recesywny | Zaburzenia pigmentacji, niedosłuch odbiorczy | Współwystępowanie choroby Hirschsprunga |
Tabela 1. Rodzaje zespołu Waardenburga. | ||||
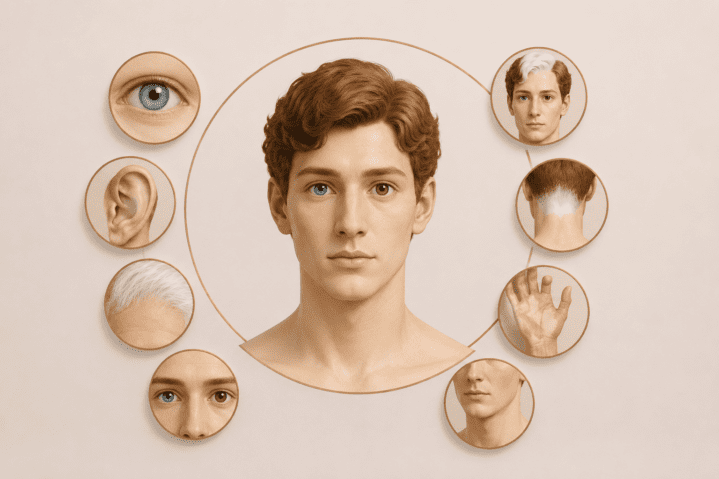 Klinicznie zespół Waardenburga często rozpoznaje się na podstawie charakterystycznych cech morfologicznych, które ujawniają się po urodzeniu. Podstawą rozpoznania jest szczegółowe badanie fizykalne i obszerny wywiad rodzinny. Typowe cechy to biała grzywka, szeroka nasada nosa oraz heterochromia tęczówek (różnica w kolorze oczu). Niektóre dzieci mogą nie przejść wstępnego badania słuchu u noworodków, jeśli zostanie ono przeprowadzone, podczas gdy inne mogą budzić obawy później z powodu braku reakcji słuchowej. Nie wszyscy pacjenci z zespołem Waardenburga wykazują wszystkie objawy kliniczne. Zmiany pigmentacyjne w zespole Waardenburga obejmują skórę, włosy i oczy. Objawy skórne mogą obejmować plamy achromiczne i hiperpigmentowane plamy nałożone na obszary o prawidłowej pigmentacji. Nieprawidłowości oczne często charakteryzują się heterochromią tęczówki lub obustronną izohipochromią. Rozpoznanie stawia się przede wszystkim na podstawie objawów klinicznych, na podstawie ustalonych kryteriów głównych i pobocznych. Rozpoznanie kliniczne zespołu Waardenburga typu 1 wymaga obecności 2 kryteriów głównych lub 1 kryterium głównego i 2 kryteriów pobocznych.
Klinicznie zespół Waardenburga często rozpoznaje się na podstawie charakterystycznych cech morfologicznych, które ujawniają się po urodzeniu. Podstawą rozpoznania jest szczegółowe badanie fizykalne i obszerny wywiad rodzinny. Typowe cechy to biała grzywka, szeroka nasada nosa oraz heterochromia tęczówek (różnica w kolorze oczu). Niektóre dzieci mogą nie przejść wstępnego badania słuchu u noworodków, jeśli zostanie ono przeprowadzone, podczas gdy inne mogą budzić obawy później z powodu braku reakcji słuchowej. Nie wszyscy pacjenci z zespołem Waardenburga wykazują wszystkie objawy kliniczne. Zmiany pigmentacyjne w zespole Waardenburga obejmują skórę, włosy i oczy. Objawy skórne mogą obejmować plamy achromiczne i hiperpigmentowane plamy nałożone na obszary o prawidłowej pigmentacji. Nieprawidłowości oczne często charakteryzują się heterochromią tęczówki lub obustronną izohipochromią. Rozpoznanie stawia się przede wszystkim na podstawie objawów klinicznych, na podstawie ustalonych kryteriów głównych i pobocznych. Rozpoznanie kliniczne zespołu Waardenburga typu 1 wymaga obecności 2 kryteriów głównych lub 1 kryterium głównego i 2 kryteriów pobocznych.
Kryteria diagnostyczne zespołu Waardenburga zostały opracowane w 1992 roku przez Waardenburg Consortium pod przewodnictwem Michael Farrer. Kryteria te odnoszą się głównie do diagnostyki zespołu Waardenburga typu 1 (WS1). Rozpoznanie można postawić przy obecności dwóch kryteriów większych lub jednego kryterium większego i dwóch mniejszych (Tabela 2.)
| Kryteria większe | Kryteria mniejsze |
| Wrodzony niedosłuch odbiorczy | Szeroka i wysoka nasada nosa |
| Zaburzenia pigmentacji tęczówek (heterochromia, hipopigmentacja, jaskrawo niebieskie tęczówki) | Wrodzone bielactwo / odbarwienia skóry |
| Biała grzywka lub hipopigmentacja włosów | Synophrys (zrośnięte lub nadmiernie zbliżone brwi) |
| Dystopia canthorum (boczne przemieszczenie przyśrodkowych kątów szpar powiekowych) | Hipoplazja skrzydełek nosa |
| Krewny pierwszego stopnia z rozpoznanym zespołem Waardenburga | Przedwczesne siwienie włosów (<30. r.ż.) |
Tabela 2. Kryteria diagnostyczne zespołu Waardenburga. | |
Zespół Waardenburga jest chorobą genetyczną, dla której nie opracowano dotychczas leczenia przyczynowego. Postępowanie terapeutyczne ma charakter objawowy i koncentruje się przede wszystkim na wczesnym rozpoznaniu oraz leczeniu zaburzeń słuchu, co ma kluczowe znaczenie dla prawidłowego rozwoju mowy, komunikacji i integracji społecznej pacjentów. W zależności od stopnia niedosłuchu stosuje się aparaty słuchowe lub implanty ślimakowe, szczególnie w przypadkach wrodzonej głuchoty.
Istotnym elementem opieki jest również profilaktyka dermatologiczna, obejmująca ochronę przeciwsłoneczną w obszarach odbarwionej skóry, bardziej podatnych na uszkodzenia promieniowaniem UV. Rokowanie dotyczące długości życia pacjentów jest zazwyczaj dobre, jednak przebieg kliniczny może być związany z licznymi powikłaniami wynikającymi z zaburzeń rozwoju tkanek pochodzących z grzebienia nerwowego. Do możliwych manifestacji należą m.in. zaburzenia okulistyczne, wady układu kostno-mięśniowego, niepełnosprawność intelektualna czy zaburzenia psychiczne. Nasilenie objawów zależy od rodzaju mutacji genetycznej, przy czym mutacje homozygotyczne oraz niektóre warianty typu 4 zwykle wiążą się z cięższym przebiegiem choroby.
Kluczową rolę w diagnostyce i prowadzeniu pacjentów odgrywa poradnictwo genetyczne, szczególnie ze względu na rodzinny charakter i autosomalnie dominujący sposób dziedziczenia większości postaci zespołu. Badania prenatalne umożliwiają identyfikację mutacji patogennych, jednak nie pozwalają na jednoznaczne przewidzenie fenotypu ani stopnia nasilenia objawów. Ze względu na wielonarządowy charakter choroby opieka nad pacjentem wymaga współpracy interdyscyplinarnego zespołu specjalistów, obejmującego genetyków, audiologów, dermatologów, okulistów oraz chirurgów.
Piśmiennictwo:
- Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34(8):656–665.
- Pingault V, Ente D, Dastot-Le Moal F, et al. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31(4):391–406.
- Song J, Feng Y, Acke FR, et al. Hearing loss in Waardenburg syndrome: a systematic review. Clin Genet. 2016;89(4):416–425.
- Farrer LA, Grundfast KM, Amos J, et al. Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: first report of the WS consortium. Am J Hum Genet. 1992;50(5):902–913.
- Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8(3):251–255.
- Baldwin CT, Hoth CF, Amos JA, et al. Identification of a locus for Waardenburg syndrome type II on chromosome 3q. Nat Genet. 1992;1(3):219–223.
- Smith SD, Kelley PM, Kenyon JB, Hoover D. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. J Med Genet. 2000;37(6):446–448.
- Bondurand N, Dastot-Le Moal F, Stanchina L, et al. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet. 2007;81(6):1169–1185.
- Reynolds JE, Meyer JM, Landa B, et al. Analysis of variability of clinical manifestations in Waardenburg syndrome. Am J Med Genet. 1995;57(4):540–547.
- Liu XZ, Newton VE, Read AP. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am J Med Genet. 1995;55(1):95–100.
- Hageman MJ. Heterogeneity in Waardenburg syndrome. Am J Hum Genet. 1977;29(5):468–485.
- Milunsky JM. Waardenburg syndrome type I and type II. GeneReviews®. University of Washington, Seattle; updated 2023.
- Tamayo ML, Gelvez N, Rodriguez M, Florez S, Varon C. Screening program for Waardenburg syndrome in Colombia: clinical definition and phenotypic variability. Am J Med Genet A. 2008;146A(8):1026–1031.
- Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int J Dermatol. 1999;38(9):656–663.
- Nayak CS, Isaacson G. Worldwide distribution of Waardenburg syndrome. Ann Otol Rhinol Laryngol. 2003;112(9 Pt 1):817–820.
- Ahmed Jan N, Muiño Mosquera L, Van Esch H, et al. Revisiting the diagnostic criteria for Waardenburg syndrome type I. Genet Med. 2022;24(4):889–897.